
被控酒駕! 比利時男子患「這罕病」被判無罪
有名比利時男子於近日被控酒駕,但經法院審理,男子竟被判無罪,其原因是因為他患有自動釀酒綜合症(ABS,auto-brewery syndrome),這是種身體會自動產生酒精的極其罕見疾病。綜合外媒報導,男子的律師表示,該名男子在一家啤酒廠工作,但這純屬巧合,他並沒有酒後開車,且經3名醫生的檢查,醫生證實他患有自動釀酒綜合症,法院宣判該名男子無罪。對此,比利時聖盧卡斯醫院(AZ Sint-Lucas)的臨床生物學家弗洛林(Lisa Florin)解釋說,患有這種疾病的人所產生的酒精,與酒精飲料中含有的酒精相同,但患者通常感覺不到酒精的影響。弗洛林補充說,人們並不是生來就患有自動釀酒綜合症,而是當他們已經患有另一種腸道相關疾病時才會患上該疾病。自動釀酒綜合症的患者可能會出現與酒精中毒類似的症狀,例如言語不清、跌倒、運動功能喪失、頭暈和打嗝等等。
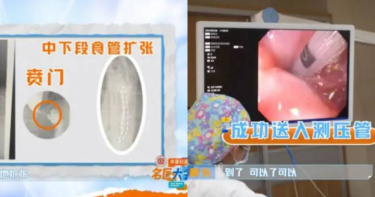
每次吃飯只能吞一粒米!他狂嘔暴瘦15公斤 才知是罹患罕見疾病
大陸江西省一名江姓男子罹患了罕見的「賁門失弛緩症」,每次吃飯只要稍微加速,喉嚨就會被噎住,尤其在吃硬食時更為嚴重,甚至會把吃進去的東西直接嘔吐出來,導致他一次只能吞下一粒米,體重也從75公斤下滑至60公斤。後來,經過相關的內視鏡手術後,切開賁門處過厚的肌肉,讓賁門括約肌徹底放鬆,約一個星期左右就可以恢復正常飲食。根據陸媒《紅星新聞》的報導,江男到上海市的醫院求診,係因大約在3年前,他發現自己無法吞食稍大的食物,有時胸口還會出現劇烈疼痛,每次吃飯能一粒一粒吞。而在醫院檢查後發現,江男吃下去的食物並沒有順著食道進到胃部,而是直接堵在了食道裡面。江男接受了胃鏡和胃食道的測壓檢查後,醫師察覺他的賁門括約肌無法正常放鬆,食道難以蠕動,讓食物卡在「門口」,且在食物的壓力下,江男的食道也出現明顯的擴張。而這樣的症狀在醫學上被稱為「賁門失弛緩症」,雖然發病率並不高,卻會讓患者感受極大的痛苦,連吃飯都變成艱鉅的任務。醫師與專家一同會診後,決定施行「經口內視鏡肌肉切開術」(POEM手術),先在江男的胃食道管壁黏膜處切出小口,利用內視鏡鑽到黏膜下,切開賁門處過厚的肌肉,讓賁門括約肌得以徹底放鬆,從源頭就解決問題。經過1個小時左右的內視鏡手術後,江男賁門處的肌肉已經順利被切開,在創口不大的情況下,他在24小時後就可以喝水,等待約一個星期左右便能恢復正常飲食。

兒子罹罕病「多毛像狼人」 菲律賓媽自責:懷孕吃貓的詛咒…醫曝真相
以為受到詛咒!菲律賓有一名媽媽,在懷孕期間吃了一隻黑貓,結果生下的兒子異常多毛,看起來就像是狼一樣,她認為這是主要原因,為此十分自責,但醫師診斷後,表示這是一種罕見疾病,僅有10億分之一的機率得到。據《每日星報》報導,來自菲律賓阿帕堯的阿爾瑪(Alma Gamongan)育有1女2子,小兒子出生時長滿了頭髮,鬢角、臉上、頸部和手臂都長滿毛髮,原本就迷信的她深信,是因為懷孕時吃了一隻野貓,而給自己帶來詛咒。阿爾瑪擔心兒子的與眾不同去學校會被欺負。(圖/翻攝自每日星報)阿爾瑪懷孕期間,她對野貓這類偏遠山區不常見的食物有著無法克制的慾望,因此從住在村裡的朋友那找來一隻黑貓,用草藥炒來吃,直到兒子出生後,她立刻後悔,並且感到內疚,而村民們也一致認為這是詛咒的結果。阿爾瑪透露,其他2個孩子都沒有這種情況,她很擔心兒子去上學,因為與眾不同遭到霸凌,「我們嘗試幫他剪掉頭髮,但只會長得更長、更粗,所以之後就不再這樣做了」。不過,有皮膚科醫師證實,男童只是罹患了極為罕見的「先天性遺傳多毛症」,又稱狼人綜合症,全世界僅有50例個案,每10億人中才有1個,而這個病症無法治癒,只能靠雷射除毛等治療緩解情況。

不能丟下他!地震瞬間抱身障同學「一起逃」 高一學生獲2萬獎學金
花蓮3日發生規模7.2強震,福建廈門也有明顯震感,廈門雙十中學的高一生張智勛在地震當下,沒有任何猶豫,馬上抱起同桌的身障同學一起逃出教室。張智勛事後表示,自己當下只有一個想法,就是不能丟下同學,要抱著對方一起跑!而他勇敢的舉動不僅獲得了表揚,也為自己贏得了人民幣5000元(約台幣2.2萬元)的獎學金。據《廈門日報》報導,張智勛就讀於廈門雙十中學高一一班,班上同學樂樂(化名)患有罕見疾病,手腳萎縮變形,只有手臂前端的一小段能夠出力,行動不便都依靠輪椅代步。因此樂樂入學之後,校方把高一班安排在一樓,導師找到張智勛,詢問他能否與樂樂同桌,平常也能幫忙照顧勒勒,他馬上就答應了。高一一班的班導師說,樂樂的母親平常會到學校幫忙,但也沒辦法整天隨時陪在身邊,樂樂母親不在時,班上的男同學就會幫忙把樂樂抱上抱下,比如說要去上實驗課,必須跨樓層,而主要出力的就是張智勛,他不在時,其他同學也會幫忙。樂樂告訴記者,地震發生的時候他感到非常難受,不知所措,因為自己沒辦法走路,也沒辦法動,但張智勛把他抱起來時,他一下子感覺「活過來了」。而張智勛則提到,地震發生那一剎那,自己沒有任何猶豫,也沒有任何空白,第一時間的反應就是,不能把樂樂丟下,要抱他一起跑。張智勛還告訴記者,樂樂曾經告訴他,樂樂國中時學校舉行的疏散演習,同學們都是抱起他一起跑的,「我一直記著這件事,印象深刻」。事件發生後,班主任將監控視頻播放給同學們看,引來全班同學的熱烈掌聲。而中國阿里巴巴公益基金會也看到了張智勛勇敢的表現,聯繫校方與廈門日報,為他頒發「阿里公益天天正能量」5000元人民幣(約2.2萬元新台幣)的獎學金。

全台首例捐腦者!漸凍人吐心願 愛妻握他手簽名遺愛人間
「台灣腦庫」去年11月於台大揭牌後,就積極準備接受病友捐腦,台中慈濟醫院50歲漸凍病人黃鼎承親身經歷罕疾身心折磨,堅決捐出腦組織,台中慈濟醫院與台灣腦庫合作,黃鼎承20日終於圓夢,成為第1例運動神經元疾病病友的捐腦入庫。黃鼎承3年前診斷罹患罕見運動神經元退化疾病「肌萎縮性脊髓側索硬化症」(俗稱「漸凍症」),他面對生命挑戰從不輕言放棄,但遍尋名醫治療與復健,身體依然快速退化,生活起居全仰賴母親與太太,兩個孩子也受到影響。台中慈濟醫院心蓮病房護理長黃美玲(左二)、醫療志工(左一)陪伴黃鼎承(右一)與黃太太(右二)到病房外吸收陽光的滋潤。(圖/台中慈濟醫院提供)黃鼎承病情發展快速到頸部以下無法動彈,僅能仰賴呼吸器維生,接受台中慈濟醫院安寧團隊居家療護時,自主表明要依照「安寧緩和醫療條例」,自主撤除維生醫療並捐出全身可用器官。台中慈濟醫院表示,黃鼎承積極搜尋漸凍人器官捐贈資訊,得知去年成立的台灣腦庫公開徵求運動神經元疾病病人腦組織,要做為醫學研究及開發新藥,親身經歷罕見疾病身心折磨與拖累的他,堅決要捐出腦組織,期盼能拯救更多家庭,心蓮安寧團隊依照囑咐聯繫台灣腦庫。台灣腦庫代表人台大醫院醫師謝松蒼南下,在病榻前向黃鼎承道謝,並保證「一定會珍惜這份國家寶藏!」他指出,黃先生的大愛會深刻影響腦神經疾病診斷,並帶來治療的成就。謝松蒼告訴黃鼎承,越來越多研究顯示腦神經疾病與周邊器官會相互影響,黃鼎承也決定願意同時捐出內臟、提供血液做次世代基因定序;最後無法執筆的黃鼎承由太太協助握筆,簽屬捐贈同意書,他激動說:「這場病生得太有意義,感謝上天賜予的恩典。」黃太太說,家人原本不諒解先生選擇撤除維生醫療的決定,如今他以身示教展現勇敢與大愛,如同麥子不死,家人以他為榮,更期盼未來能拯救更多罕見疾病的病人與家庭。

13歲少女患罕病成弱勢! 奇美「罕病中心」介入給予幫助
13歲小美因患罕見疾病導致下肢功能受影響與脊椎側彎,行動需輪椅代步,又為單親的經濟弱勢家庭,小美父親一直苦撐著照顧和經濟上的雙重壓力。然而,奇美醫院罕見疾病照護團隊的介入,協助整合社會補助資源源,幫忙加入罕見疾病基金會,並連結家扶中心及民間社福團體提供急難補助。不僅提供了醫療上的協助,更在社會和心理層面上給予全方位的支持。罕見疾病中心協助患者 榮獲SNQ國家品質標章奇美醫院遺傳暨罕見疾病中心講座教授林秀娟說明,由於罕見疾病種類繁多,各類疾病照護需求差異很大,需有結合各層面的團隊整合服務。奇美醫院以個案管理模式提供罕見疾病病人與其家屬充分之疾病資訊及心理支持,並提供照護諮詢及生育關懷等服務,2023年榮獲國家生技醫療產業策進會(生策會)頒發SNQ國家品質標章認證。奇美醫院為擴及遺傳性及罕見疾病之照護,2013年成立遺傳諮詢中心,同與成大醫院為雲嘉南地區惟二通過國民健康署認證之遺傳諮詢中心一。為加強罕見疾病照護,2022年更名為遺傳暨罕見疾病中心,並建立跨科部及跨院「遺傳及罕見疾病服務網絡」,服務包括:(1)提供遺傳諮詢、檢驗轉診,協助追蹤及預防。(2)提供國內外遺傳診斷、治療之轉診及轉檢聯繫。(3)推動遺傳罕見疾病診斷、諮詢及診療發展與研究,及(4)推動遺傳及罕見疾病相關社會教育。「罕見疾病全人照護模式」 跨科部整合、跨院提供服務林秀娟指出,儘管服務對象多為其他醫院通報的個案,而且居住地涵蓋臺南、雲林、嘉義等地區,執行上雖為困難,但奇美醫院藉由跨科部組成「罕見疾病照護服務」執行團隊,以核心工作小組為聯絡窗口,整合醫療、長照、社福等各方面資源,努力讓罕見疾病病人及家庭都能得到全方位的照護服務。五全照護概念 持續醫療與社區支持林秀娟表示,我國對於罕見疾病照護相當努力並已有良好成果,但仍存在醫療資源集中於急重症治療為主的醫學中心,科際整合及全人照護仍待加強。病友離開大醫院後,在地照護的可近性與連續性仍不足,有待結合醫療、長照、社福體系及社區等在地資源形成支持網絡。為此,奇美醫院建構完整的罕見疾病照護網絡,利用五全(全人、全程、全家、全隊、全社區)照護概念,檢視罕見疾病照護現況,連結醫療、長照、社福及社區資源,提供罕病家庭連續性完整性的全人照護,不僅使病人得到持續的醫療照護,也能在社區中獲得相應的支持。

仲恩生醫35元登興櫃漲逾51% 董座:幹細胞藥今年將申請日本藥證
仲恩生醫(7729)上周五(8日)召開興櫃前法人說明會,於今(11日)以每股35元登錄興櫃,今早開盤股價上衝至53元,漲幅逾51%,隨後漲幅收斂至7.5%。董事長暨總經理王玲美表示,仲恩長期專注神經罕見疾病藥物研發,目前進展最快的是治療小腦萎縮症(SCA)的Stemchymal幹細胞藥物,預估將在2024至2025年提出日本藥證申請。仲恩生醫創立於2007年,致力於脂肪間質幹細胞投入罕見疾病療法藥物研發,期望為罕病病友提供有效治療方案,研發至今已累積多項創新幹細胞技術平台開發、前臨床及臨床試驗,開發適應症涵蓋小腦萎縮症、亨丁頓舞蹈症兩項罕病,以及急性發炎性疾病包含急性缺血性腦中風、急性肝衰竭、急性呼吸窘迫症等。仲恩生醫副總柯景懷進一步指出,經由動物實驗及離體細胞實驗證實,Stemchymal治療機制可透過特定的分泌因子,經由自體吞噬的路徑,達到減少異常蛋白質堆疊、調節發炎反應、抗氧化能力、支持神經細胞修復、生長;從動物行為實驗上可看到顯著的改善。目前市面上研發藥物多為單一靶點,柯景懷表示,而Stemchymal可多靶點全方位達到保護神經細胞及抑制突變蛋白質的堆積,明顯優於其他競爭對手。目前仲恩已與日本Reprocell、韓國SCM Lifescience 兩家公司達成Stemchymal技術授權,同時,仲恩也持續積極拓展其他國際市場如美國及歐洲等,爭取技轉授權到海外的機會。在藥證取得方面,仲恩生醫除日本市場預計在2024、2025年提出藥證申請外,台灣方面則可望在「再生醫療製劑條例」通過後,依「有附款許可」取得暫時性許可。搭配衛福部已於今年1月施行「已於醫藥先進國核准之罕見疾病藥品認定併查驗登記審查試辦方案」,兩種途徑都可能縮短Stemchymal在台上市時程,為台灣罕病病友提供治療方案,未來也將繼續專注臨床需求,以孤兒藥為目標,持續開發其他適應症。

爽挖耳朵10年突「耳痛流膿」 檢查竟罹癌了
大陸一名50多歲周姓男子喜歡隨手拿起挖耳勺掏耳朵,習慣持續了將近10年,直到最近,左耳痛、耳流膿讓他不得不就醫,這才發現罹患癌症,所幸經過手術切除,並輔以放化療,恢復良好。醫師說明,長期掏耳朵刺激了外耳道的表皮,引起皮膚滲出,惡性循環使得耳道裡長出新生物,誘發外耳道癌。根據陸媒《中國新聞網》報導,南方醫科大學珠江醫院耳鼻喉頭頸外科中心主任張宏徵表示,外耳道癌是一種少見的惡性腫瘤,約佔頭頸部腫瘤的0.2%,整體發生率約1/100萬人,屬於罕見疾病,容易漏診,確診時往往已屬於局部晚期。他提到,外耳道癌臨床表現不典型,初期易誤診為外耳道炎或中耳炎,主要臨床表現為耳流膿、耳痛、聽力下降、耳鳴、耳悶、眩暈等症狀;晚期腫瘤因侵犯範圍擴大,可出現相應顏面神經及後組顱神經麻痺症狀,如顏面神經麻痺、聲音沙啞、飲水嗆咳、吞嚥困難等,侵犯顳顎關節可出現張口受限。張宏徵指出,外耳道癌的確診要靠病理,同時還需要進行聽力學、顳骨CT、顳骨頸部增強MRI、肺部CT等相關檢查,腫瘤累及大血管時還需做MRA、MRV,PET-CT可以幫助判斷是否出現遠處轉移。張宏徵進一步指出,外耳道癌多見於40歲至60歲的成人,和多種高危險因子相關,其中包括反覆的上皮刺激,例如習慣性挖耳、長期的慢性化膿性中耳炎和外耳道炎均有誘發外耳道癌的風險;另外,在亞洲人群中,頭頸部放射治療是外耳道鱗狀細胞癌重要的誘發因素,經過放射治療後的鼻咽癌患者,外耳道鱗狀細胞癌的發生率約0.15%,較健康者高出1000倍。張宏徵稱,外耳道癌主要分為鱗狀細胞癌、腺樣囊性癌、基底細胞癌;鱗狀細胞癌最常見,佔40%至60%,侵襲性較強且預後最差;腺樣囊性癌約佔40%,雖生長緩慢,但容易復發及遠端轉移;基底細胞癌佔5 %至10%,手術後效果優於其它病理類型。張宏徵解釋,外耳道癌的治療以手術切除為主,同時標靶治療和免疫治療也可以作為很好的補充;而外耳道癌的預後一般與分期、病理類型相關,早期外耳道癌手術切除後預後較好,5年存活率可達到90%-100%,晚期僅35.8%-72.5%,因此,保持良好的生活習慣,盡量避免外耳道癌的危險因子,有相關症狀時及時就診,做到早發現、早期診斷、早治療。
用AI技術「復活」已故父親安慰91歲奶奶 他鼻酸感嘆:老人不能受刺激
大陸遼寧省一名男子擔心奶奶受到刺激,便沒有誠實告訴她父親已過世的消息,但奶奶實在太想念兒子,於是他就使用了AI技術來「復活」爸爸,藉此讓她再次透過手機見到對方。根據陸媒《瀟湘晨報》的報導,孫姓男子提到,奶奶今年已經91歲,心臟健康不佳、身體狀況也堪憂,而父親罹患了罕見疾病,並在去年5月離開人世,從發現到逝世只過了不到11個月。不過,因為不想讓奶奶擔心,孫男只能瞞著她,謊稱爸爸正在外地治療,不方便一起返鄉。孫男表示,春節獨自去看奶奶的時候,奶奶一直詢問著父親的狀況,想要親自打電話與他聯繫,而孫男只能趕緊編出理由,說醫院檢查時身上不能帶著金屬,也無法使用手機,奶奶才特別拜託孫子,希望他到醫院探望父親時,能夠請對方簡單錄影和她說句話。後來,孫男被一部含有AI情節的電影所啟發,決定下載付費軟體,用自拍鏡頭自己錄製一段影片後,將父親的樣貌融合上去。起初孫男順利拍攝完畢後,才想起父親沒有蓄鬍,遂趕緊刮掉鬍子重錄,但因為自己也太過於思念爸爸,他在錄影時仍忍不住數次掉下眼淚,「錄影對我來說是巨大的壓力,我也很想父親,接下來還會不會用這種形式來安慰奶奶,我也說不清楚」。孫男感嘆,奶奶是個特別堅強的人,只要繼續瞞著她,跟她說爸爸的狀況良好,奶奶就不會往壞處想,「他就像一棵大樹一樣照顧家裡所有的人…只能走一步算一步了」。而這段紀錄被上傳至網路後,就有不少網友頗感共鳴,留言道「他AI換臉扮演的是奶奶思念的兒子,又何嘗不是自己剛剛失去的懷念的父親」。
創世界首例!日本男童接受3親人「活體器官捐贈」 移植成功已順利出院
日本京都大學醫學部附屬醫院4日宣布,一位患先天性疾病的10歲男童接受雙親及阿公捐的活體肺與肝移植手術成功,同時也成為全球首例,已順利於3月1日出院。根據日媒報導,2023年11月時一名未滿10歲的男童因為罹患「先天性角化不全症」,接受40多歲雙親捐的左右肺一部分及60歲阿公提供的一部分肝臟。手術期間共用了4間手術室,時間長達18小時以上,約30名醫師參與,最後手術順利完成,男童及其雙親、阿公也已經在3月1日平安出院,捐贈的3名親人也都恢復正常生活。京都大附醫表示,在國外有少數案例是同時移植腦死患者提供的肺與肝,日本死後捐贈器官者少,沒有相關案例。由健康的人提供部分器官且同時移植成功是全世界首例,未來將有擴大新的治療可能性。事實上,受贈器官的男孩因為染色體異常,除了有先天性角化不全症的罕見疾病外,他曾在2歲時因為再生不良性貧血,4歲時就曾接受過妹妹捐贈的骨髓做移植手術,但之後肺與肝也發生狀況,因此必須再次接受移植手術。

29歲美女作家「瞎書」驚傳逝世! 生前罹罕病體重不足40kg 曾嘆:一定要吃三餐
大陸知名的美女作家「瞎書」因罹患罕見疾病「脊髓空洞症」,於近日驚傳逝世,享年29歲。「瞎書」曾向網友透露,因工作忙碌常常熬夜,三餐不正常,工作時一天甚至只吃一餐,即便病得很嚴重,高燒39度還熬夜工作,瞎書還曾寫道「大家一定不要學我啊!一定要認真,健康吃三餐,好好睡覺,多鍛鍊!」而患病後的她暴瘦,一度不到40公斤,她近年頻繁進出醫院與病魔抗爭,如今還是不幸離世,令人不勝唏噓。據《星島頭條》報導,「瞎書」在生前創作多部暢銷作品,包括《我不會喜歡你》、《楞次定律》、《一顆青蘋果》、《巴洛克複調》、《時止之年》等,她擅長以女性視角,宣揚男女平等、獨立又互相扶持的愛情觀。創作中,則以2019年發表的《我不會喜歡你》最受推崇。「瞎書」曾向網友透露,她工作時一天只吃一餐飯,又常吃外賣,加上經常熬夜寫文,即便病得很嚴重,高燒39度她還熬夜工作, 瞎書還曾寫道「大家一定不要學我啊!一定要認真,健康吃三餐,好好睡覺,多鍛鍊!」到了2021年5月她在通宵寫作後發高燒,之後病情惡化,出現心絞痛、胃痛等問題,最後被確認罹患基因缺憾,所引起的脊髓空洞症罕見疾病。發病後,「瞎書」的體重一度由50公斤暴跌至39.55公斤,暴瘦如柴,去年6月「瞎書」肺炎高燒不退,至9月因肺部感染嚴重,醫院第2次下達病危通知,經搶救後情況慢慢好轉,「瞎書」還曾發文向讀者報平安,不過,到了今年2月,還是不幸離世,令人不勝唏噓。「瞎書」罹患罕見疾病「脊髓空洞症」。(圖/微博)「瞎書」生前的生活習慣,引起熱議,網友紛紛留言指,年輕時也要維持健康的作息習慣,外賣雖然方便,但不能依賴。脊髓空洞症是種非常罕見且不易診斷的疾病,可分為先天性和後天性,後天性多因外傷、發炎、腫瘤和脊椎側彎引起。其症狀包括頭痛、四肢和背痛、肌肉萎縮、肌肉痙攣、感覺喪失、平衡感變差和脊椎側彎等,接著會出現感覺喪失、大小便失禁,該疾病無法根治,大多只能防止病情持續惡化。

廣西婦1年確診新冠4次…竟罹「百萬分之一罕病」 常見症狀曝光
即使疫情趨緩,但新冠肺炎仍存在,許多民眾就多次確診。中國廣西有一名女子,在一年內染疫4次,最後檢查出「Good's症候群」(免疫缺乏合併胸腺瘤),是一種發病率只有百萬分之一的罕見疾病,也是反覆確診的根源。據《南國早報》報導,柳州54歲婦人今年第1天確診,這是她4度染疫,光是2023年已經中獎3次,再度確診讓她超崩潰,自從去年1月開始,就出現咳嗽、咳痰,她曾5度在當地醫院住院,每1、2個月就被診斷出新型冠狀病毒感染,雖然治療後就轉陰,但出院不久症狀又來了。婦人短短一年「4陽」,決定找出原因,檢查發現免疫力很低,醫師詢問病史後,發現她在2年前曾接受胸腺瘤手術,半年來還曾罹患口腔扁平苔蘚、鼻竇炎,經常腹瀉,體重也日漸下降。醫師將這些症狀連結在一起,發現與Good's症候群符合。據了解,這是一種罕見的自身免疫缺陷疾病,主要特徵是胸腺瘤和低丙種球蛋白血症,發病率只有1.5/100萬,由於疾病的罕見性與隱匿性,容易發生誤診和漏診,患者最終因嚴重的感染而死亡。經過嚴謹的病例討論,婦人確診為Good's症候群。截至2024年2月18日,是中國首例反覆感染新冠病毒的Good's症候群患者,醫護團隊主要針對其免疫力制定治療方案,10多天後情況完全好轉並出院,後續還需長期每月注射丙種球蛋白增強機體免疫力。

《我和我的冠軍女兒》要角罹患罕見疾病不幸逝世 年僅19歲
印度知名勵志電影《我和我的冠軍女兒》於2016年上映,這部由印度男星阿米爾罕領銜主演的電影在全球爆紅,甚至締造全球3億美元的票房紀錄。而如今有消息指出,其中飾演次女芭碧塔的演員巴特納加爾(Suhani Bhatnagar)因罹患罕見疾病猝逝,享年19歲。根據印度媒體報導指出,巴特納加爾是在2023年底突然發生手部腫脹的情況,後來逐漸蔓延至全身。中間當地醫生一直檢查,卻查無原因。直到今年2月,巴特納加爾才由德里的全印度醫學科學研究所 (AIIMS) 的醫師診斷出是罹患罕見的肌肉炎症皮肌炎。據了解,皮肌炎雖被認為是一種自體免疫疾病,但目前尚無切確的發病原因,該病會引起皮膚、肌肉發炎,同時也會導致皮疹、腫脹和肌肉無力。除了骨骼肌之外,心臟、胃腸道、肺部和關節亦可能受到疾病的侵犯。報導中指出,巴特納加爾在確診肌肉炎症皮肌炎之後,有接受類固醇的治療,但自身免疫系統受到嚴重影響,後續又疑似因為腿部摔傷骨折的因素,結果進而引發嚴重的併發症。雖然醫師有想辦法治療,但最終仍不敵病情,病逝於全印度醫學科學研究所,享年19歲。

王志安道歉並捐百萬日圓 罕病基金會婉拒:已完成退款
大陸央視前記者王志安日前在脫口秀節目嘲諷罕病律師陳俊翰,引發軒然大波,而陳俊翰11日驟逝,王志安在社群平台發出沒來得及送達的道歉信,並透露捐出百萬日圓給罕病基金會。罕病基金會今天晚間發布聲明表示,接受其善意婉拒捐款,已完成退款。罕病基金會表示,已退回大陸央視前記者王志安的百萬日圓捐款,並在官網公開退款匯水單。(圖/罕病基金會提供)王志安日前在網路脫口秀節目《賀瓏夜夜秀》中,疑似影射民進黨為選舉利用身障律師陳俊翰做秀,引發抨擊。沒想到陳俊翰卻於春節期間離世,享年40歲。王志安15日發出道歉信表示,「驚聞陳律師去世,我的歉意再也無法抵達,真是令人無限的懊悔和遺憾」,並表示決定為台灣的罕見病基金會捐款100萬日元,「錢雖然不多,也算是我誠懇道歉的佐證吧。」罕病基金會表示,本會於農曆年假前獲台北富邦銀行通知,有一筆透過日本公司TOKYOFUKOU CO.,LTD捐款日幣100萬元(約合台幣21萬)。經網路搜尋得知王志安先生為該公司(東京風行株式會社)創會人,且日前透過自媒體頻道宣稱捐款100萬日幣給台灣罕見疾病基金會,以及後續王先生所發佈之道歉聲明文中亦提及此筆捐款。罕病基金會表示,經董事會決議,本會應無立場代表台灣120萬身心障礙者及陳俊翰律師本人接受王先生道歉,而且無法認同侵犯尊嚴者及其道歉與捐助款項之間產生對價關聯,因此婉拒王志安先生支捐款,並已完成退款。罕病基金會也在協會官網公開退款匯水單。罕病基金會表示,陳俊翰律師代表全台120萬身心障礙朋友,長期投身身障者及罕病族群權益倡議,全盡畢生心力伸張身心障礙者人權,眾人萬分欽佩俊翰律師的無比勇氣、智慧與努力,除萬分不捨陳俊翰律師驟逝,本會更將持續發揚俊翰律師的信念及理想,與台灣社福公益各界,共同為更進步更美好的台灣社會戮力不懈。

台版「非常律師」陳俊翰過世 曾因SMA失去求生意志…棄高薪工作回台原因曝
位列民進黨不分區立委提名人的律師陳俊翰,兒時罹患脊髓性肌肉萎縮症(SMA)致使行動不便,而他先前因被脫口秀節目《賀瓏夜夜秀》的來賓、前中國大陸央視記者王志安於節目中談論,受到外界更多關注,怎料今(15)日傳出過世的消息,隨後民進黨證實,陳俊翰已於11日凌晨逝世,享年40歲。其實陳俊翰的病情一度讓他失去求生意志,好在母親謝季珍的陪伴在側,讓他決定堅持下去,後來更是放棄原本的美國高薪工作機會,回台推動罕見疾病及身心障礙相關政策,幫助弱勢群體發聲,勵志的生命經驗也讓他被外界封為台版「非常律師」。根據《ETtoday新聞雲》報導,陳俊翰早在1歲左右發現罹患脊髓性肌肉萎縮症,此病終將造成運動神經元喪失功能,進而肌肉無力、萎縮;在他就讀小六年級病情急轉直下,後來還被送進加護病房急救長達2個月,期間一度喪失求生意志,告訴母親「不要救我,我真的撐不下去了」,後來也因母親的長久陪伴,讓他及時轉念,有了堅持下去的動力。儘管陳俊翰失去雙腿,身體僅剩頭部、幾根手指頭還能自主活動,還遭告知若不用藥,存活時間僅剩3年至4年,但他高中畢業後仍考上台灣大學,雙主修會計系與法律系的課程,之後2度赴美取得法學博士學位,順利通過考試取得律師執照。不過陳俊翰選擇放棄原本的美國高薪工作機會、較為優越的治療環境回台,進入進入中研院法研所,也在政府部門推動罕見疾病及身心障礙相關政策,今年更是第11屆立法委員選舉,民進黨不分區名單中的身障提名人。他說,美國保險的機制強調「無差別、無歧視」,待在美國可以得到更為妥善的照顧與治療,反觀台灣卻有多達數十種罕病用藥尚未給付,因此盼能透過自身力量推動相關權益,「每一條生命都該被珍惜。」陳俊翰並稱,他覺得自己能有機會實現個人理想已經非常幸運,希望未來的台灣,所有的障礙者都不必受到先天條件的限制,「哪怕讓多一點人有機會接受用藥,也會比我自己在國外獲得治療更有價值。」後來長期鑽研人權、身心障礙政策與法律的他,更被譽為台版「非常律師」、「生命鬥士」。如今陳俊翰過世的消息一出,民進黨也以聲明回應,「陳俊翰律師因疑似感冒引起併發症,2/10前往台大醫院新竹分院急診,雖經醫護人員極力搶救,仍於2/11凌晨不幸逝世。我們聞訊感到無比震驚與悲傷,俊翰的家人低調辦理後事,年節期間未對外公布,今日委由民進黨黨中央代為發布消息。感謝各界關心,也請給家屬空間處理相關事宜。」