罕病
」 罕病 罕見疾病 醫師 健保 疾病
台灣長照3.0藏千億缺口? 蔡壁如:可借鏡日本長照保險制度
根據日本總務省統計局的統計,日本65歲老年人口已經佔總人口的29.4%,根據日本的資料指出,2025年每5人有1位高齡失智症,高齡化加上失智者多,導致長照費用支出增多,在台灣也有相同現象,為了因應超高齡化社來臨,台灣在2026年推出長照3.0,前立委蔡壁如指出,日本的長照保險制度,採取全民參與方式,規定所有年滿40歲的國民都必須參加並繳納長照保險費,台灣長照3.0以「稅收」制為主2026年的預算達到新台幣千億元是史上新高,若以日本長照保險規模約健保五分之一來看,台灣長照要達全覆蓋經費恐須二千億元,等於仍有一千多億元的缺口,因此,長照3.0想要落地化,財源穩定是考驗。財政、人力培育到系統完善 蔡壁如:台灣須借鏡「長照可不是砸錢就沒事,更需仰賴完善的人力與制度規劃,務實地提出可行方案,真正為需要者帶來福祉,不然口號喊久了只是政治口水戰,徒留社會內耗。」蔡壁如直言,日本的政策是規定所有年滿40歲的國民都必須參加並繳納長照保險費,長照保險費的金額會依據各地方自治的財政狀況及長照需求,每3年重新檢討與調整保險費的徵收制度設計,強調社會共同承擔照護責任,同時也希望透過合理分攤,減輕家庭個別負擔,確保長者能依需求獲得適切照護服務。反觀國內,目前仍靠遺贈稅、菸酒稅等「稅收」支應,其核心理念同樣在於建構社區整合式服務體系,強調在地老化、社區照護與多元服務選擇,並逐步朝全民參與與合理分攤費用的方向努力,從日本經驗來看,台灣長照3.0的千億投入若要產生實質效果,可借鏡其保險制度的穩定財源設計,盡量壓低公家支出,提高使用者費用,降低入住機構比例,以家庭照顧為優先,在地終老,在宅臨終大力推動,以及強調社會責任分擔,進一步提升制度的可近性與永續發展。另一方面,日本在長照服務內容、專業人力培育及社會支持系統的完善,也能作為台灣優化長照3.0的重要參考,從「單純補貼服務」轉向「專業長期照顧」,讓更多長者能夠在熟悉的社區安心老化,減少家庭壓力,提升整體社會福祉,才能有效應對超高齡社會的挑戰。「老」不是罪也非悲情 活得好還是「長」值得深思過年期間看了一本描述日本長照「長日將盡,來杯Sake吧」。日本是亞洲高齡化趨勢最快的國家,六十歲是青老,七十歲是中老,八十歲是老老。超高齡社會的問題,執政當局都著重在財政、醫療層面。但是,社會層面非常複雜也很重要。社會問題包括:「一個人的老後,單身、老老、失智者的照顧,偏鄉老人無人送終,都是老人國的憂鬱。」另外,老人國的老人認為;老不是原罪,也不悲情。即使老,還是要有生活品質(Quality of life),即使有共病症還是要有老伴,要交女朋友。即使腰酸背痛,五十肩,還是要早睡早起,努力克服,讓生活起居按年輕的步調前進!在活得好或活得長哪一個比較重要?其實沒有標準答案,因為我們的習俗對長壽是一種恭維,民族性對談論死亡是禁忌。好死不如歹活?好死不如賴活?毫無意義的延長生命的治療,浪費醫療,無效醫療,對長照都是一大考驗。更多醫健新聞報導「國際罕病日」⎪罕病SMA病友治療後找回生活品質 呼籲各界重視罕病!備孕從飲食開始做起!把握飲食「4關鍵」、補充必備「5種營養素」,迎接好孕到來!

眼皮下垂別輕忽!輕熟女誤當疲勞 惡化就醫確診罕病
眼皮下垂小心是身體給的警訊!大陸浙江一名30歲輕熟女長時間感到眼皮沉重,原以為只是工作過勞,但雙眼逐漸無法正常睜開,還出現視線重影的狀況,才迅速就醫。經檢查被確診是罕病「重症肌無力」,所幸在醫療團隊長達2年的規範治療下,輕熟女慢慢恢復正常生活,視力與一般人沒太大區別。根據陸媒《縱覽新聞》報導,浙江大學醫學院附屬第一醫院近日分享一起案例,有一名30歲輕熟女患者自2年多前,出現眼瞼下垂症狀,平時努力睜眼整個人還是無精打采,隨後情況還惡化,看東西有重影。師診斷後發現,其臨床表現具有明顯的「晨輕暮重」特徵,即早晨起床時狀態尚可,但隨時間推移,疲勞感會導致症狀加劇,這正是罕病重症肌無力典型的臨床表現。經過醫療團隊長達2年的規範治療,輕熟女患者的眼睛問題獲得改善,生活上與一般人沒太大區別。醫師指出,重症肌無力是一種自體免疫疾病,特別提醒民眾,若發現眼皮出現波動性下垂、複視、肢體無力,甚至伴隨胸悶或呼吸困難,應立即就醫檢查。由於該病初期容易被誤認為疲勞或老化,若能早期診斷並進行醫療介入,能有效緩解症狀並大幅提升生活品質。至於重症肌無力在台灣的現行治療方式,台灣神經免疫醫學會理事長葉建宏曾表示,有包括俗稱「大力丸」的抗乙醯膽鹼酵素劑、類固醇、快速調節免疫機能的血漿置換術(俗稱「換血」)與靜脈注射免疫球蛋白,以及胸腺切除手術等。

夫妻無家族遺傳史「卻檢出聽損基因」 醫:宜孕前篩檢找出隱性遺傳風險
32歲的林小姐與先生新婚不久,雙方家族皆無明顯的遺傳病史,自認健康狀況良好。在計畫懷孕前,夫妻倆到奇美醫院婦產部主治醫師徐英倫門診諮詢,在徐醫師建議下進行「擴展性帶因者篩檢」。報告卻意外顯示,夫妻兩人皆為聽損基因(GJB2)的帶因者,而未來寶寶將有25%的機率罹患先天性聽力障礙。林小姐起初相當震驚,但在醫療團隊的說明與協助下,轉診至生殖醫學中心,選擇透過胚胎著床前基因診斷(PGD)技術,成功篩選出不帶致病基因的健康胚胎進行植入,目前已順利懷孕20週,產檢狀況一切正常,胎兒發育良好。家族沒人生病也可能有遺傳風險 抽血可篩檢隱性遺傳疾病徐英倫醫師說明,根據2024年發表的臺灣本土基因研究顯示,即便家族中沒有人生病,遺傳帶因現象在臺灣仍比想像中普遍。臺灣族群中,約每五人就有一人(21.6%)帶有聽損基因變異;此外,其他在臺灣常見的隱性遺傳疾病還包括甲型/乙型地中海貧血、威爾森氏症(影響肝臟與神經)及龐貝氏症(影響肌肉功能)等。這些疾病的帶因者通常外觀健康,但若夫妻雙方都帶有同一致病基因,將有四分之一的機率誕下病童。隨著基因定序技術的進步,現代醫學的擴展性帶因者篩檢,準父母可藉由一次抽血,篩檢數百種隱性遺傳疾病,替下一代多一層保障。擴展性帶因者篩檢預先找出隱性遺傳風險 再決定生育計畫徐英倫醫師指出,美國婦產科醫學會(ACOG)指引建議,無論種族或家族史,所有計畫懷孕或已懷孕的女性,皆可考慮接受擴展性帶因者篩檢。研究顯示,當篩檢發現夫妻雙方皆為同一致病基因帶因者時,若疾病後果嚴重(如會影響壽命或生活自理),大多數夫妻會因此改變生育計畫。根據臨床數據指出,在懷孕前得知高風險結果的夫妻中,約有62%會選擇透過試管嬰兒合併胚胎診斷(IVF+PGD)或產前診斷的方式,來預防遺憾發生。擴展性帶因者篩檢並非製造焦慮 而是能做好更多準備徐英倫醫師強調,擴展性帶因者篩檢並非要製造焦慮,而是為了讓未來的父母有更充分的準備。建議所有計畫懷孕的夫妻,特別是希望降低罕見疾病風險的家庭,可至婦產科門診諮詢。透過科學的檢測與專業的遺傳諮詢,基因檢測能將隱形的遺傳風險具象化,讓每個寶寶都能在最完善的規劃下健康誕生。【延伸閱讀】新手爸天生僅有三根手指!北醫阻斷「罕病遺傳基因」迎健康新生兒備孕爸媽看過來!PGT-H基因篩檢 有助評估下一代健康風險https://www.healthnews.com.tw/readnews.php?id=67187
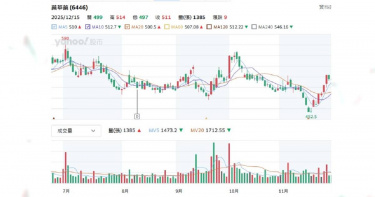
新藥股大漲跌1/這二檔營收利多股價亮眼 「它」納入中國目錄成亮點
台股力拼站回28K,AI概念股仍是主旋律,大數據分析等科技導入生技醫療產業也加速新藥研發進程,臨床試驗數據一翻兩瞪眼好壞在眼前,CTWANT調查2025年股價亮眼上漲與慘跌共四檔新藥股,2026年展望是個個有希望,最關鍵的還是營收財報給證據。 先來看看最新一例,2025年12月7日中國國家醫保局公布首版《商業健康保險創新藥品目錄》,納入18家企業19款高值創新藥,聚焦CAR-T、雙抗、罕見病及阿茲海默症等超出基本醫保範圍的產品,研發PTS肺癌新藥的共信-KY(6617)子公司天津紅日健達康醫藥科技公告「治療中央型氣道阻塞產品-PTS302」,成為台股唯一上榜個股,消息曝光,股價從78.8元上漲到86.6元,旋即又短暫下跌86.4元,再逐步漲到15日收盤價90.5元,到這周連四跌、周五收盤價85元。 CTWANT調查,共信12月3日公布11月合併營收259.1萬元,年減12.5%,累計雖年增11.42%,但投資人擔憂目錄效應需時間轉化實際訂單。 至2025年12月15日止,觀察仁新醫藥(6696)、藥華藥(6446)個股股價變化,其均以創新藥為核心,近期受臨床進展、營運利多驅動,股價有所表現。仁新醫藥近一月從270元上漲到最高442元,近三月則從162元漲到周五收盤407元;藥華藥近一月從453元漲到最高519元,近三月則最高漲到559元。仁新醫藥專注罕見疾病新藥,子公司Belite Bio(美國納斯達克BLTE)Tinlarebant三期臨床於2025年12月1日公布,針對青少年斯特格病變臨床三期試驗(DRAGON)解盲之關鍵性數據亮眼,共收案104位青少年受試者,並成功達成主要療效指標。Belite董事長暨執行長林雨新醫學博士表示,「DRAGON最終試驗結果讓斯特格病變疾病治療迎來了歷史性突破,Tinlarebant將成為首個治療這種具威脅性疾病的潛在新藥,期盼藉此帶給無藥可醫的患者及其家屬治療新希望。」BLTE股價從今年初低點62.09美元,漲到近期的最高點156美元,「三期解盲成最大催化劑。」證券分析師表示。Belite規劃於2026年上半年與各國監管機構討論Tinlarebant的潛在後續計畫與新藥查驗登記申請(NDA)送件事宜,並向美國FDA遞交新藥查驗登記申請(NDA)。Belite宣布Tinlarebant抑制視網膜萎縮進展達預期,股價飆至新高,仁新持股 43.94%間接受惠,募資126億元不折價增資當日完成。仁新2026年關鍵為Tinlarebant FDA核准,預計上半年送NDA。藥華藥向美國FDA申請Ropeg新適應症「原發性血小板過多症(ET)」,目標2026上半年取證。圖左起為執行長林國鐘、董事長詹青柳、總經理黃正谷。(圖/CTWANT資料照)藥華藥旗下真性紅血球增多症(PV)新藥Ropeginterferon alfa-2b(Ropeg,P1101)用於新適應症罕病血癌藥銷售勁揚,股價高檔震盪,10月底完成向美國FDA申請Ropeg新適應症「原發性血小板過多症(ET)」送件,若順利獲准,最快有望於2026年上半年取得美國ET藥證,成為公司啟動第二成長曲線的重要動能。藥華藥11月營收15.5億元,月增10.63%,年增53.02%,已連續3個月刷新單月歷史新高;累計前11個月達137.11億元,較去年同期的86.02億元,成長59.38%;前三季獲利達35.69億元,年增翻倍,每股盈餘9.66元,賺近一個股本。根據CTWANT調查,藥華藥也正同時研發新款癌症藥物。此外,Ropeg筆型注射劑的補充性生物製劑查驗登記申請(sBLA)審查亦同步進行中。FDA已通知審查目標完成日(PDUFA date)為2026年2月14日。
ADCC缺乏症治療納健保惹議 王婉諭喊生命無價:罕病不該效益論
時代力量黨主席王婉諭今(2)日表示,近期有醫師將ADCC缺乏症(芳香族L-胺基酸類脫羧基酶缺乏症)每針新台幣近1億元基因治療花費由健保來扛的政策,講成是「剜肉補瘡」排擠別的資源,這種說法是太殘酷,畢竟罕病是基因纏上門,根本無從預防,健保本來就需要承擔這類風險,且生命無價,對罕病家庭來講,多活一天都是最寶貴的饋贈。罕見疾病AADC缺乏症是基因突變的體染色體隱性遺傳疾病,這會影響多巴胺和血清素的製造,進一步影響運動控制、情緒調節、內分泌及心血管功能等多個生理系統。據了解,若父母雙方皆帶有異常基因,則子女有四分之一機率患病,而台灣ADCC缺乏症發生率比全球高得多,台灣大概每3萬名新生兒中即有1人罹患此病。時代力量黨主席王婉諭(圖)長期關心罕病家庭。(圖/周志龍攝)此外,據臨床文獻記載,由於ADCC缺乏症症狀多樣又容易被誤診成腦性麻痺等其他問題,因此診斷需仰賴醫師經驗與警覺性,如有必要會透過腰椎穿刺抽取腦脊髓液,檢驗其中神經傳導物質及其代謝產物的含量比例以確診,目前這項篩檢也已納入新生兒篩檢項目範疇。 儘管過去ADCC缺乏症幾乎無藥可醫,讓病童多數難以活超過5歲,但近年由臺大醫院團隊研發出一種創新的基因替代療法,透過立體定位神經外科手術將功能性基因包裹在腺病毒載體中,接著直接注射到大腦特定的「豆狀核」區域,讓患者體內能開始重新合成多巴胺,並逐步達成正常運動發展。這項罕病藥物技術要價近億元,並已於2025年12月起納入台灣健保,限縮適用範圍在18個月以上至6歲以下的患者。ADCC缺乏症治療納入健保是否有當,各界爭論不休。圖為藥房示意,無指涉當事人。(示意圖/黃耀徵攝)然而,網紅胸腔科醫師蘇一峰則不以為然地指出,健保12月起決定花費龐大預算,讓患者壽命從用藥前平均7歲壽命延長到20多歲餘命,而且新藥並非完全能治癒疾病,患者注射後仍需要一輩子醫療照顧,健保有限資源還可能排擠到更多病患的健保資源,此話一出引起「救命」與「浪費」兩派舌戰。王婉諭對此表示,AADC缺乏症這種基因問題除非父母特地篩檢,不然根本無法預防,這樣的罕病正是健保存在的原因,多少罕病家屬明知有解藥,卻因天價治療費無力負擔,只能眼睜睜看著孩子在眼前病故,那種無力感將壓垮一個家庭,如今要用效益估計,何其殘忍。王婉諭強調,健保問題根源在於不敢正視財務結構失衡,加上「二代健保補充保費」規則既不公平也不永續,如今要拿特定藥品補助獵巫,根本無濟於事,若不全面改革開源,未來醫療體系的崩壞只是早晚的事。

12月多項新制上路 TPASS升級回饋30%、iPASS MONEY要另下載APP
2024年12月起,多項政策與制度陸續上路,涵蓋交通、健保、數位支付與長者福利等領域。其中,交通部升級推出TPASS 2.0+方案,國道客運最高享30%回饋;健保新增單劑要價達1億元的罕病用藥;iPASS MONEY平台服務轉移至專屬App;臺北市敬老卡則擴大應用範圍,年底前可用於泡溫泉。【TPASS 2.0+優惠擴大】交通部自今年1月推動TPASS 2.0公共運輸常客優惠方案,每月搭乘公共運具滿11次可享最多30%回饋。12月1日起升級為TPASS 2.0+,凡搭乘國內91條中長途國道客運路線,每月搭2次可獲15%回饋、4次以上可享30%回饋,其他運具則維持原有條件。【臺鐵調整車型並加開班次】臺鐵公司宣布,自114年12月23日起調整部分列車車型與班次。彰化-花蓮間的278次與285次列車將改為EMU3000型自強號,樹林-花蓮間224次與235次則調整為普悠瑪。另新增臺中-潮州間EMU3000型自強號191與196次列車,以及彰化-竹南間下午區間車2班,提供更彈性運能。【健保納入高價罕病用藥】健保署公告,自12月起新增3項健保藥品給付,涵蓋轉移性胰臟癌、停經後轉移性乳癌及芳香族L-胺基酸脫羧酶缺乏症(AADC)藥物。其中AADC為罕病新藥,單劑藥價高達1億元,預估首年約有13人使用,整體藥費高達13億元。【iPASS MONEY服務調整】一卡通公司公告,自2025年12月3日起,LINE Pay錢包中的iPASS MONEY僅保留查詢餘額、提領、好友轉帳與帳戶紀錄查詢等功能。若欲使用儲值、轉帳、消費等完整功能,民眾需另行下載iPASS MONEY專屬App操作。【北市敬老卡點數可泡湯】臺北市政府擴大敬老卡使用範圍,年底前卡友可使用點數於特約業者折抵泡湯費用,目前已有約20家業者參與。至於是否可擴及至KTV使用,因涉及產業管理層面,社會局仍在研議可行性。

吃藥一劑要1億!12月六大新制上路 交通、醫療、氣象全面調整
進入12月,民眾生活與交通相關的多項政策新制陸續實施,涵蓋運輸優惠、醫療補助、氣象預警及地方交通等面向。《CTWANT》整理出六大變革重點,協助讀者快速掌握最新規範內容。TPASS 2.0公共運輸常客優惠12月上路,最高可享30%搭乘回饋。(圖/報系資料照)一、TPASS 2.0+搭乘回饋再升級交通部公路局自12月1日起推出「TPASS 2.0+」公共運輸常客優惠方案。凡每月搭乘91條中長途國道客運路線達2次者,可獲15%乘車金額回饋;搭乘4次以上,回饋升至30%,連續假期亦可適用。詳細規則可至交通部官網查詢。二、台鐵12月23日改點 調整111列次台鐵將於12月23日實施列車時刻微調,共計111列車受影響,其中15班列車變動幅度逾5分鐘。本次改點重點包括:‧ 彰化-花蓮普悠瑪改為EMU3000型自強號‧ 台中-潮州增開2班EMU3000型列車(191次、196次)‧ 海線午後增開區間車2班次,改善尖峰運能時刻表可於台鐵公司官網與「台鐵e訂通」App查詢。三、強風特報鄉鎮等級制正式上線中央氣象署推出的「陸上強風特報鄉鎮燈號」於12月1日正式實施,透過黃、橙、紅三種燈號區分強風等級,民眾可更快速掌握鄉鎮層級風力預警,及早因應防範。四、1億元AADC基因治療藥納健保 創史上單價新高衛福部最新公告中,三項藥品自12月起納入健保給付,包括治療轉移性胰腺癌、乳癌與罕病AADC(芳香族L-胺基酸類脫羧基酶缺乏症)。其中AADC基因治療藥物單劑高達1億元,為健保史上單價最高藥品。五、台中交通罰單新增超商繳費通路台中市警局宣布,自12月1日起,行人違規、慢車違規及道路障礙等罰單,除可至警察局與郵局繳納,亦新增全國四大超商繳費選項,提升便民服務效率。六、雲林西螺大橋封閉維修 封橋期長達一年雲林縣政府表示,西螺大橋將自12月1日上午8點起封橋進行補強工程,預計至2027年1月4日止,期間全橋封閉。用路人請改道台1線溪州大橋並遵守交通管制指引。陸上強風特報鄉鎮燈號12月起正式實施,採黃、橙、紅三等級警示。(圖/中央氣象署提供)

禁婚擬放寬到四等親? 醫師搖頭:生畸形兒機率恐飆
立法院法制局建議,將近親結婚限制從六等親放寬至四等親,研議修正民法限縮禁婚親等。不孕症治療權威醫師強調「萬萬不可」,近親通婚除了有倫理問題,寶寶更容易有先天疾病、畸形風險,進而增加健保負擔,優生學仍有其必要性。我國民國87年修正民法,基於優生學考量,將旁系血親禁婚範圍訂為六親等以內。立法院法制局研析報告指出,近年多數國家皆立法縮小禁婚範圍,包括旁系血親禁婚不超出三親等,因此研議修正民法限縮禁婚親等,以符社會現況。報告指出,現代社會家庭結構變遷,親戚關係日漸疏遠,年輕世代鮮少與親戚往來,可能在不知情的情況下誤觸近親禁婚規定,導致婚姻無效。隨著醫療科技進步,只要在孕期確實進行產檢,就可大幅降低產下罕病兒機率。茂盛醫院院長李茂盛表示,立法院擬修法,將近親結婚限制從六等親放寬至四等親,是「萬萬不可」,除了有倫理問題,過去也發現,表兄妹間通婚生子,會出現很多畸形兒、先天疾病,對台灣社會不好,更會浪費健保資源。李茂盛說,雖然現今有基因檢測,但也不是100%精準,萬一寶寶生下來有先天疾病,整個家庭也受影響,每個人都得不到幸福,「優生學有其必要」。針對放寬是否有助減緩少子化?李茂盛認為,「一點幫助都沒有」,少子化的問題並不在近親通婚,而是政府應加強托育政策、補助計畫,鼓勵年輕人早婚等等。立法應該長遠、周全,而非變來變去,否則只會對台灣社會造成更多紛亂。

罹罕病靠「輸液續命」嚴重骨鬆 25歲女申請安樂死:讓我走吧
澳洲25歲女子荷蘭(Annaliese Holland),因長年飽受罕見且末期的神經疾病折磨,近日決定啟動澳洲合法的「自願協助死亡」(VAD)程序,希望能「以自己的方式告別生命」。她的故事在當地社會引發廣泛討論。《紐約郵報》(New York Post)報導,荷蘭自童年起便反覆進出醫院,長期受慢性疼痛、噁心及持續嘔吐困擾。醫師多年來無法確診,使她的病情在青少年時期急速惡化。她最終被診斷為「自體免疫性交感神經節病變」,這是一種罕見疾病,患者的免疫系統會攻擊控制呼吸、心跳、腸胃等不自主生理功能的神經節。確診前,荷蘭腸胃功能幾乎癱瘓,無法消化食物。餵食管治療完全無效,她持續嘔吐,醫師最終只能以完全靜脈營養(TPN)維持生命。這種治療風險極高,只要感染便可能迅速引發敗血症。荷蘭透露,她已歷經多達25次敗血症,生死一線。醫師在荷蘭18歲時將其轉入成人醫院後才找出病因,但當她22歲時,病情已被判定為末期。多年藥物治療也導致她骨質脆弱,出現嚴重骨質疏鬆,造成多處脊椎骨折與胸骨骨折,並對心肺帶來危險壓力。她說,雖然生命中仍有美好時刻,但每天都在忍受「無法形容的劇痛」。長期病痛讓她的青春歲月幾乎全被醫院佔據,像是18歲與21歲生日都在病房度過,而同齡朋友已開始結婚、組家庭。她無奈地說,「大家的人生都在前進,而我被困住了。我不是在生活,只是在勉強活著。」在得知生命終將走向終點後,荷蘭決定選擇以VAD結束痛苦,並向醫療團隊表明不願再承受無止境的折磨。家人起初難以接受,尤其是父親曾問「妳要放棄嗎?」但在某次她被醫師從瀕死邊緣救回後,她含淚求父親「讓我走吧」,父親這才理解女兒已承受太多。經3週評估後,荷蘭獲准使用VAD。得知批准當下,她忍不住落淚,「那是一種奇怪的喜悅。因為我終於能在痛苦與平靜之間做出選擇。」她坦言,雖然自己即將解脫,但痛苦卻落在家人心上,她將盡力安排好一切,減少對親人的傷害,「選擇VAD不是放棄,而是在盡全力與病魔對抗後,終於能勇敢地說一句:我受夠了。」

男青春期1年內身高198cm長到223cm 身體不適就醫確診罕病
有不少人希望自己長得高,但因為生病的關係長太高就不是件好事!大陸浙江一名20歲劉姓男子小時候不高,在青春期時身高急速飆升,甚至曾在1年內猛長25公分,目前他的身高已經有223公分。後來劉男常常有體力不支、身體不適的情形,就醫後才發現確診罕見疾病「垂體性巨人症」。根據陸媒《陝視新聞》報導,來自浙江溫州的劉男因為身高突出、目前是223公分,他在求學時期曾被送往體校受訓打籃球,但訓練時常感到體力不支、易疲倦,就醫後初步檢查發現血糖水平極高,每天需注射80單位胰島素控制。之後劉男又進一步檢測,結果更顯示他體內生長激素分泌超標高達300多倍,醫師最終確診為「垂體良性腫瘤」、合併「肢端肥大症」、「垂體性巨人症」及「糖尿病」。溫州醫科大學第一附屬醫院指出,若生長激素過量分泌發生在青少年時期,會導致「巨人症」,而若發生在成年後,則容易引起「肢端肥大症」,導致手指變粗、五官變形、下顎突出等外貌改變。這類疾病不僅影響外觀,還可能導致呼吸系統、心血管、消化及代謝等多器官併發症,包括關節疼痛、頭痛、代謝異常,甚至提高腫瘤風險,如結腸瘜肉或甲狀腺結節。劉男經手術治療後,體內激素水平已恢復正常,血糖與其他指標均受控制,目前已康復出院。院方神經外科主任蘇志鵬提醒,正常孩童每年長高約5至7公分,青春期最多10公分,若發現孩子短時間內異常暴高,應及早就醫檢查,以免延誤治療時機;另外,醫師也呼籲,家長若發現孩子出現「長太快」、「手腳異常粗大」等徵兆,應提高警覺,因「巨人症」若延誤治療,恐影響一生健康。

世界上真的有天使!車臣女孩因罕病擁有雪白長髮與異色雙瞳,美貌震撼全球
一頭自然雪白的長髮、配上一藍一棕的雙眼,這位來自車臣的女孩阿米娜(Amina Ependieva)最近又讓網路熱議起來!其實這組照片拍攝於2020年,當時她才11歲,卻因獨特外貌被形容「像童話走出來」。這系列照片由俄羅斯攝影師Amina Arsakova拍下,他在街上偶遇阿米娜,被她的氣質吸住目光,於是邀請拍攝。畫面中,阿米娜只是安靜站著或坐著,沒有特別擺姿勢,卻讓人一下就記住了她——乾淨、特別、帶著難以忽視的存在感。(圖/取自AminaArsakova IG)阿米娜患有白化症,讓她的皮膚和髮色幾乎沒有黑色素;同時她也有虹膜異色症,雙眼呈現不同顏色。一些網友看到後感嘆「這根本是真實版的奇幻角色」,也有人留言「第一次看到這樣的眼睛,真的會被吸住」。(圖/取自AminaArsakova IG)這組照片再次被翻出後,討論焦點除了她的外貌,也延伸到「多樣性美感」,許多留言都提到:每個人的特質都是獨一無二的,而這樣的特別值得被看見、被尊重。(圖/取自AminaArsakova IG)四年前的畫面至今仍反覆被分享,證明這樣的純粹魅力不會被時間洗淡。她的故事也提醒大家,美不一定要符合既定框架,有些人的光,是安靜但很難忽略的那種。

23歲女星演完第一部劇突傳死訊!從童星到遺作只隔13天 伊莎貝兒泰特罹罕病辭世
美國新生代演員伊莎貝兒泰特(Isabelle Tate)於10月19日病逝,得年23歲。她生前罹患罕見的遺傳性神經疾病「進行性神經性腓骨肌萎縮症(Charcot-Marie-Tooth),消息由其所屬經紀公司Kim McCray透過社群平台證實,並表示全體同仁與家人對此深感哀痛。泰特在《9-1-1:納許維爾》中飾演角色Julie,該劇為她首次以成人身分接演作品。(圖/翻攝自Hulu)根據《Hollywood Reporter》與《Variety》報導,泰特自童年時期便參與演出與模特兒工作,後來從中田納西州立大學(Middle Tennessee State University)畢業,取得企業管理學位。畢業後,她重返演藝圈,並順利通過試鏡,獲得影集《9-1-1:納許維爾》(9-1-1: Nashville)的角色機會。泰特於6月完成該劇首集拍攝,並於10月6日正式播出。她飾演名為Julie的角色,劇中是一名坐輪椅的女性角色,出現在脫衣舞表演場景中,與Hunter McVey飾演的角色Blue互動。不料節目播出僅13天後,她便因病離世,令人不勝唏噓。她的家人在公開聲明中提到:「伊莎貝兒是一名充滿活力的鬥士,從未讓自己的身體狀況成為藉口。她用行動證明,任何限制都無法抹滅她對夢想的堅持與熱情。」經紀人McCray也在社群平台哀悼表示:「我認識她從她還是青少年開始,她最近重新投入演藝事業。《9-1-1:納許維爾》是她試鏡的第一部劇集,就成功獲得角色。她對於這次拍攝過程非常投入,也度過了難忘的時光。」Charcot-Marie-Tooth病是一種會影響神經傳導的遺傳性疾病,導致四肢肌肉逐漸萎縮、感覺喪失等慢性症狀,對日常生活造成顯著挑戰。泰特的家屬呼籲外界,若想表達哀思,可透過捐款支持Charcot-Marie-Tooth協會(CMTA),以延續泰特的愛與精神。泰特生於田納西州納許維爾,生前熱愛音樂創作與探訪動物收容所。她的家人包括母親Katerina Kazakos Tate、繼父Vishnu Jayamohan、父親John Daniel Tate及妹妹Daniella Tate。她的告別式預定於10月24日在布倫特伍德舉行。

28歲女罹患罕病終身癱瘓 冷血丈夫騙賣婚房捲款失蹤!下場曝
大陸一名28歲女子婚後,因罹患一種中樞神經系統的罕見疾病,不幸終身癱瘓,卻慘遭丈夫捲款失蹤,甚至放話「寧可坐牢也不回來」。經檢警強力介入,女子的丈夫最終因遺棄罪入獄服刑,而她在檢方協助下順利完成離婚訴訟,重獲新生。根據陸媒《荔枝新聞》報導,來自江蘇的28歲女子林靜(化名)原本是一名導遊,婚後第4年突發罕見中樞神經疾病,導致終身癱瘓,下半生要倚靠輪椅生活。當她最需要照顧時,她的丈夫常姓男子卻哄騙她出售婚房,捲走百萬元後人間蒸發。走投無路的林靜,只能向南京江北新區法院提起訴訟,指控丈夫遺棄。2021年時法院在審理過程中將案件線索移送檢察機關,承辦檢察官楊晴發現警方遲未立案,隨即要求說明,並推動偵辦。約7天後,警方正式立案同時發布通緝,常男潛逃近5年,終於在2022年底落網;2024年3月檢方以涉嫌遺棄罪提起公訴,法院判處常男10個月有期徒刑。不過案件並未就此結束,檢方發現林靜生活困難,便啟動司法救助,市、區兩級檢察院合計撥付6.5萬元(約新台幣28.2萬元)救助金,也有多個單位陸續投入協助。然而林靜仍有一個心願,就是與常男離婚,她表示,「丈夫寧可坐牢,也不願盡責。」檢方知道林靜的心願,立即提起附帶民事訴訟,林靜的離婚案得以展開;在2025年3月6日一場特殊庭審在常州監獄舉行,歷時近3個小時後,法院裁准離婚。林靜輕聲道出心聲,「婚離掉了……謝謝。」對此,檢察官楊晴感慨,「看到她重新燃起希望,一切付出都值得。」而江蘇省檢察院主管則指出,本案不僅追究刑責,更透過救助與民事訴訟,形成跨部門合作,成功解決民眾的困難。
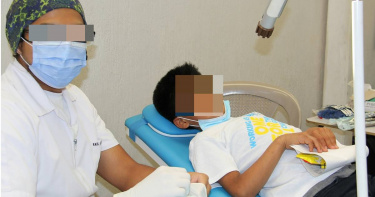
醫誤判流感!美國6歲童患罕病險喪命 母靠Google救回孩子
美國德州一名6歲男童原本因頭暈與頭痛送醫,卻被誤診為流感,結果不到一天,他便陷入無法走路、說話,甚至無法自主呼吸的險境,最後失去意識,確診為罕病「海綿狀血管瘤」。她的母親在絕望下在Google搜尋資料,意外找到專治此症的醫師,最終挽回孩子的性命。綜合《紐約郵報》、《國家廣播公司》(NBC)報導,男童威頓(Witten Daniel)今年4月因頭暈與頭痛就醫,初經初步診斷為流感,卻在入院不到24小時病情急速惡化,開始無法行走、說話、自主呼吸,最後甚至失去意識,醫師雖緊急插管維持呼吸,但仍查不出病因,後來才發現是他腦部出現罕見的「海綿狀血管瘤(Cavernous Malformation)」因腫瘤破裂出血,才導致抽搐、中風接連發生。醫師甚至一度警告威頓的母親凱西(Casey Daniel),她的兒子就算存活,也可能終身臥床並依賴呼吸器與胃管。凱西回憶,自己當時「看到孩子變成那樣,真的沒有言語能形容」。但她不放棄,透過Google搜尋資料,找到休士頓德州大學健康科學中心神經外科醫師莫柯斯(Jacques Morcos)的文章,立刻發送郵件向對方求救,並很快獲得對方回覆,對方要求她將威頓緊急轉院。所幸威頓經莫柯斯與小兒神經外科醫師沙阿(Manish Shah)的4小時手術後奇蹟般甦醒,不僅能自行呼吸,還能再度說話。6周後,他順利回到拉巴克(Lubbock)的家中,並迎來7歲生日。「我想感謝莫柯斯醫師和沙阿醫師,讓我能再見到我的朋友。」威頓在受訪時形容,他的康復過程「很美好」。如今他已經重返校園就讀小學二年級,甚至準備再次站上棒球場,事實上,在確診海綿狀血管瘤前,他還被評為少年棒球聯盟球隊的MVP。專家指出,約每500人就有1人帶有海綿狀血管畸形,但大多數患者沒有症狀,常見於20至40歲才發病,症狀包括癲癇、出血,或頭痛、視力模糊或言語障礙等神經系統問題,約20%病例與遺傳有關。

66歲黃仲崑曝健康狀況 坦言「罹罕病」左手不停顫抖
黃仲崑與「金曲小姐」洪小喬今(12日)同台亮相,為11月22日即將登場的《臺北大巨蛋 民歌大團圓》演唱會站台。除分享音樂心情,66歲黃仲崑也透露父親當年因帕金森氏症過世,而自己在拿到敬老卡後,經檢查竟被診斷出同樣的症狀。黃仲崑笑說第一時間就把消息告訴兒子黃遠:「嘿嘿,你的DNA應該跟我一樣!」他淡然表示:「人生到這個階段就很坦然,現在永遠是右手拿麥克風,左手不能拿就換手,沒有什麼不能做的,看山就是山,看水就是水。」黃仲崑(左)、洪小喬一起為11月12日即將登場的《臺北大巨蛋 民歌大團圓》演唱會站台。(圖/趙文彬攝)不過,他的太太黃露蕙(Lulu)則補充,由於不同醫生意見不一,有醫師認為黃仲崑是「原發性震顫」而非帕金森氏症,且他還自告奮勇參加人體實驗,結果因神經系統代償作用,讓左手開始抖動。她打趣虧老公:「他就這張臉值錢,要出來賺錢給我花。」目前黃仲崑固定每月回診,也透過中醫調理身體。黃仲崑坦言曾動過腦部手術,復原後近來又出現左手顫抖,但他態度樂觀,認為年紀、基因帶來的病變都應該坦然面對。他也笑談與小自己17歲的Lulu感情,當年結婚時被外界說是「父女戀」,如今反而變成「姊弟戀」,夫妻倆互動仍舊甜蜜,還笑稱老婆Lulu「什麼事都要管我」,展現牢不可破的感情。

星國5歲女童罹罕病!台男3000公里捐骨髓 手寫信打氣「祝福妳健康長大」
人間處處有溫情!新加坡一名5歲女童罹患罕見貧血病,遠在3000公里以外的台灣,有男子以2萬分之1的機率配對成功,大方捐贈骨髓進行移植,還附上手寫信替她打氣,「經歷這次捐贈過程,才知道生病是多麼辛苦,每次進醫院的妳一定更辛苦,很幸運能和妳配對成功……祝福妳未來能健康長大」,相當感人。根據《聯合早報》報導,女童林子馨和媽媽俞建芬昨(7日)出席「骨髓捐贈計劃」(BMDP)組織舉辦的活動,分享尋求骨髓捐獻者的經歷。林子馨去年8月罹患罕見的再生不良性貧血,骨髓不能產生足夠的新細胞補充血液細胞,醫師建議3個月內接受骨髓移殖,只是父母和哥哥都不適合捐獻。好在約莫1個月後迎來好消息,對方是一名來自台灣的男子。不過,林子馨必須在移植手術前接受化療,意味著可能會經歷嘔吐、疼痛和失去胃口等副作用,也必須剃光頭以減少感染風險。由於從小就把長髮視為寶貝,也愛打扮成公主,俞建芬擔心女兒化療要剃光頭會難過,出乎意料的是,當天不僅沒哭鬧,還對她微笑,讓她感到無比驕傲。林子馨順利結束1小時的手術,但接下來的2個月仍要留院觀察。按照規定,捐獻者和受益者須等至少2年才能見面。俞建芬說:「我們只知道他是一名30多歲的台灣男子。我們有回信感謝他,並說希望明年能到台灣見他」。

愛到生命盡頭!罕病女孩拒絕兩次求婚 26歲前男友猝逝留遺書與積蓄助她圓夢
大陸重慶一名22歲女孩胡心瑤(筆名鹿靈瑤)自14歲起與罕見疾病抗爭多年,歷經數十次病危,卻依舊努力生活。8月31日,她收到一封來自前男友的遺書與5萬元人民幣(約新台幣22萬元)轉帳,才得知這位曾向她兩度求婚的年輕輔警(公安輔助警務人員),已因急性重症胰腺炎搶救無效去世,年僅26歲。分手後,肖先生匿名資助胡心瑤,直至去世後才被揭曉身份。根據《封面新聞》與《觀世記》報導,胡心瑤的病史漫長而艱辛。2018年,她確診IgA血管炎,先後併發腎衰竭、心衰竭、消化道出血與骨頭壞死,靠著打工與社會捐助支撐高額醫療費。她為自己取筆名「鹿靈瑤」,象徵在精神世界裡仍能奔跑的自由靈魂。2024年1月,她透過親友介紹認識了來自內蒙古、在重慶擔任輔警的肖先生。對方明知她的病情,仍選擇與她交往,並在短短兩個月內兩度求婚,但胡心瑤因不想拖累他而拒絕,甚至提出分手。儘管如此,肖先生始終默默守護,化名「愛心網友」匿名捐助,並在工作之餘兼職多份工作,累計資助她超過8萬元人民幣(約新台幣35萬元)。肖先生的遺體在成都火化,胡心瑤捧花送行,完成最後告別。今年8月,胡心瑤舉行一場「嫁給自己」的婚禮,象徵面對病痛仍要堅定活下去,並宣布計畫成立「病友小家」,希望凝聚相似處境的患者互相扶持。然而8月底,她卻迎來沉重打擊。肖先生因病猝逝,臨終前委託友人將積蓄與遺書交給她。8月31日,胡心瑤收到前男友的遺書與5萬元積蓄(約新台幣22萬元),用以支持她的「病友小家」計畫。遺書中,肖先生寫道:「我知道,建一個『病友小家』是你一直放在心上的夢想,我想把這筆錢都資助給你。答應我,無論以後有多難,都要替我好好看這個世界。」胡心瑤收到消息後悲痛不已,並主動聯繫肖先生的父母,表示願意將錢歸還。但對方父母堅持尊重兒子的遺願,支持她完成計畫。她說:「從那一刻起,我已經把他的父母當成我的父母,只要我還在一天,就會盡力照顧他們。」目前,「鹿靈瑤病友之家」已找到合適場地,270平方米的房子年租金僅5500元人民幣(約新台幣2.4萬元),遠低於市場行情。計畫於10月底正式開放,已有40多名病友報名參與。屆時將安排醫師進行健康講座,並由心理師提供免費輔導,同時設有交流休閒空間。胡心瑤表示,這份延續愛的支持,將成為她推動病友互助平台的最大動力。分手後,肖先生匿名資助胡心瑤,直至去世後才被揭曉身份。

媽分享小一罕病女兒開學被質疑「不好好產檢」 婦科名醫:不必傷口撒鹽
一名媽媽近日分享小一罕病女兒要開學了,而因為女兒有狄蘭吉氏症,被網友酸她為何不做產檢。對此,婦產科名醫蘇怡寧指出,該種罕病即使做羊膜穿刺檢查也一樣,「在別人的傷口上撒鹽,真的大可不必。」媽媽在Threads發文,她在懷孕過程有產檢,醫師沒有建議做羊膜穿刺,其餘都做了,「我女兒這罕病是基因的突變,現在醫學非常的發達,已經可以檢驗出來了。」蘇怡寧在臉書發文回應,狄蘭氏症候群(Cornelia de Lange syndrome)是一種罕見的基因異常疾病,大多數病例都是偶發,沒有家族史可追蹤,「新生兒發生率大約是萬分之一到五萬分之一,臨床表現涉及身體外觀發育智力及多系統器官,雖然目前無法治癒,但透過跨專業的早期介入與支持性治療,的確能顯著改善生活品質。」蘇怡寧表示,該疾病在產前真的非常難診斷,即使做羊膜穿刺染色體檢查或是基因晶片也沒辦法檢測出,「唯一可能的機會,是在超音波發現異常時,再透過全基因定序才有可能找到答案,但實務上太困難了。」對於網友問媽媽「為什麼產檢不好好做」,蘇怡寧說,在別人的傷口上撒鹽大可不必,這只是顯露無知,在一個稱最美麗風景是人的國家,有人拿孩子的弱點來取笑,有教養的人不是應該申出援手嗎,「弱勢,不是他們的選擇,但是否展現同理心,卻是你我的選擇。」

全球2000次核試遺害至今 「下風者」家庭癌症、罕病纏身仍待補償正義
從80年前展開核子實驗開始,在1945年至1996年間,全球共進行超過2000次核試驗,其留下的健康與環境陰影至今未散。成長於1950至1960年代美國猶他州鹽湖城(Salt Lake City, Utah)的迪克森(Mary Dickson)回憶,小學時期被教導核戰來臨要「趴下掩護」;她心裡卻明白「這根本救不了我們。」多年後才發現,美國就在鄰近的內華達州進行試爆,而她所居之處正位於「下風區」。迪克森後來罹患甲狀腺癌,姐姐因紅斑性狼瘡早逝,妹妹近日被診斷腸癌轉移,姪女們也接連出現健康問題。她統計,童年住處方圓五個街區內,竟有54人出現癌症、自體免疫疾病、先天缺陷或流產。根據《CNN》報導,醫界普遍認為輻射暴露會提高癌症風險。美國環境保護署(EPA)明確指出「輻射暴露會增加罹癌機率,且劑量越高風險越大。」居於內華達試驗場周邊的亞利桑那州、內華達州、猶他州、奧勒岡州、華盛頓州與愛達荷州居民,統稱為「下風者(downwinders)」。迪克森感嘆「對我們來說,冷戰從未結束,我們仍生活在它的影響之下。」自1945年廣島與長崎遭原子彈轟炸、約11萬人當場喪生後,美蘇展開冷戰核軍備競賽,英國、法國與中國也陸續投入。1996年前,全球核試次數已逾2000次;21世紀以來,僅北韓於2017年進行過核試。史蒂文斯理工學院副教授威勒斯坦(Alex Wellerstein)形容早期決策心態「國家安全被視為絕對必要,其他風險常被忽視。」報導中提到,多數核試選址於偏遠或殖民地。美國主要在內華達與馬紹爾群島;蘇聯則在哈薩克與北冰洋新地群島;英國於澳洲與基里蒂馬蒂環礁;法國在阿爾及利亞與法屬玻里尼西亞;中國則在新疆羅布泊。哈薩克塞米巴拉金斯克試驗場1949年至1989年間進行逾450次核爆。哈薩克倡議者塞伊特諾娃(Aigerim Seitenova)指出,許多居民多年不知全貌,她親眼見家族成員在壯年接連病逝,成年後才理解與核試的關聯。她拍攝紀錄片,探討核遺產對女性的跨世代影響,並在廣島放映,凸顯馬紹爾群島、玻里尼西亞與澳洲也共享相似經驗。美國國家癌症研究所(NCI)1997年估算,1951至1962年間內華達大氣層試爆,可能造成11300至212000例額外甲狀腺癌;後續審查認為實際數字較接近低端。在哈薩克塞米巴拉金斯克,核試最密集年代的癌症與嬰兒死亡率均高於其他地區。卡內基國際和平基金會學者卡塞諾娃(Togzhan Kassenova)說「這不是過去的問題,而是仍在持續的代價。」馬紹爾群島承受的影響尤其巨大。美國1946至1958年間在當地實施67次核試,總當量相當於7232枚廣島原子彈。1954年的「Castle Bravo」試驗讓放射性落塵覆蓋朗格拉普與艾林吉奈環礁,居民癌症風險急遽上升。大量居民被迫遷離,部分在1970與1980年代嘗試返鄉卻未能持續。如今,數千名馬紹爾人移居美國本土延續社群。哥倫比亞大學研究員休斯(Ivana Nikolić Hughes)解釋,銫-137會沿食物鏈生物累積,從椰子到椰子蟹再到人類。美方在部分區域清理後污染下降,但核試設施建設與後續處理仍改變當地生態,魯尼特穹頂(Runit Dome)的安全性至今備受質疑。其實美國自1990年設立《輻射暴露補償法》(RECA)後,已支付逾27000名下風者、金額超過13億美元,並於今年延長且擴大補償範圍。哈薩克納入約120萬人補助;馬紹爾群島則認為美方補償不足。法國於2010年才承認阿爾及利亞與玻里尼西亞受害,至2021年僅半數申請者獲賠;馬克宏總統曾坦言「法國對該地負有債務」。英國則要求核試老兵依戰爭退休金制度申請,但退伍軍人團體仍爭取專門補償。從廣島與長崎的浩劫至今80年,最密集的核試已過數十載,然而亞利桑那沙漠、塞米巴拉金斯克草原與太平洋環礁,至今仍回響著當年的爆炸。迪克森說,她朋友中太多人癌症復發,那種無法消散的陰影,是看見身體任何腫塊或疼痛時的惶惑「會不會,它又回來了?」

罕病AADC基因治療「價格近億」納健保採採分期支付 首年估13人受惠
AADC缺乏症是一種遺傳性罕見疾病,通常在嬰兒早期出現症狀。由台大醫院團隊研發的AADC基因治療,2022年獲歐盟上市許可,價格近億元。健保署藥物共擬會今天(21)審查通過健保給付,將和廠商啟動價量協議,預計最慢今年11月開始給付,預估第一年13名患者受惠。芳香族L-胺基酸脫羧基酶缺乏症 (AADC)是先天基因缺損,通常在嬰兒早期出現症狀,導致嚴重的發展遲緩、動眼危象、低張力及四肢運動異常、自律神經系統失調、睡眠問題及情緒障礙等。AADC基因治療是2010年由台大醫院基因醫學部提出「AADC治療計畫」,並陸續獲得AADC缺乏症病友會、罕見疾病基金會、長榮集團張榮發總裁及生技醫藥國家型科技計畫(NRPB)的支持贊助,之後技轉給美國藥廠。歐洲藥品管理局(EMA)於2022年核准這項基因治療藥物上市。健保署副署長龐一鳴證實,今天共擬會通過AADC基因治療納入健保給付,並授權健保署和廠商進行「價量協議」,牌價近億元,實際價格會低一些。共擬會專家認為,由於藥物價格高,採「分期支付」,觀察病人穩定有效才會給付。依據國健署統計至今年7月, AADC罕病通報72人、死亡27人、存活45人。而台灣新生兒發生率約三萬分之一,高於其他國家,如日本約16萬2千分之一、美國約6萬4至9萬分之一、歐盟約11萬6千分之一。龐一鳴表示,此次通過給付的重點不在於價格多貴,這類罕病病童若沒有幫忙就活不久,國家應該盡量照顧,且相對其他國家,台灣病人數較多,早期研發跟技術都是以台灣為基地,再向國際推廣,健保更應該要支持。龐一鳴說明,依照標準流程為2個月,最慢今年11月開始給付,預估給付後第一年使用人數13人。他轉述健保署長石崇良的話,「全台灣每一個人也只付不到5元,可以救一個罕病小朋友,絕對值得!」